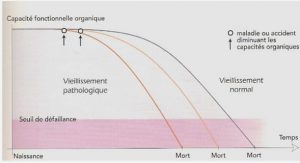
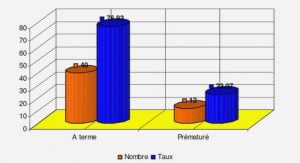
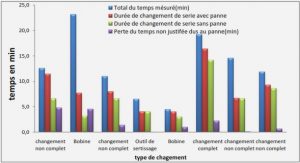
Grands principes de la recherche clinique
Historique de la recherche clinique
Certains attribuent le début de la recherche moderne sous forme d’essais clinique en double ou simple aveugle au groupe de chercheurs du Michigan en 1931, ou à ceux de l’hôpital de Londres en 1933, mais selon Kaptchuk [1] , une évaluation en aveugle avec intervention fictive aurait été réalisée lors d’une mission d’une commission royale française sur la folie du mesmérisme dans les années 1780. Cette commission, dirigée par Benjamin Franklin, alorsembassadeur américain en France, aurait été l’initiatrice de l’essai clinique contrôlé par placebo en simple aveugle. Aucune autre référence antérieure à cette méthode d’analyse des thérapies médicales n’aurait été mise en évidence. Les termes « test en aveugle » et « test en double aveugle » apparaissent pour la première fois dans des études publiées respectivement en 1935 et 1950. [2] Concernant la valeur statistique de ce type d’études, l’assignation aléatoire de sujets à un groupe de traitement ou à un groupe de contrôle (placebo) permettant des comparaisons statistiques valides entre les deux groupes à un certain niveau de confiance définissable a été décrit par Fisher [3], statisticien qui décrira donc la valeur « p », couramment utilisée actuellement. Green conclut, après ces rappels historiques, que « L’essai clinique randomisé en simple ou double aveugle contrôlé par placebo reste la méthode la plus fiable pour évaluer les bienfaits revendiqués de toute intervention thérapeutique. Bien qu’il puisse être difficile de concevoir et de mener de telles enquêtes dans certains contextes, tous les efforts doivent être faits en ce sens. » .
Encadrement légal de la recherche
A l’échelle internationale, le respect des bonnes pratiques cliniques (BPC) est un gage de qualité auquel se référent les autorités de santé des pays acceptant cette norme (l’Union européenne, le Japon et les Etats-Unis). Les BPC sont définies par la Conférence internationale d’harmonisation (ICH) comme étant la « norme concernant la conception, la conduite, l’efficacité, le suivi et l’audit des essais cliniques ainsi que l’enregistrement, l’analyse et la présentation des données s’y rattachant et qui garantit que ,
– Les données et les résultats présentés sont fiables et exacts
– Les droits, l’intégrité et l’identité des sujets de l’essai sont protégés. » .
Les principes éthiques prônés dans ces BPC trouvent leur fondement dans la déclaration d’Helsinki, rédigée pour la première fois en 1964 par l’Association médicale mondiale et relative à la recherche médicale portant sur les êtres humains. En France, la règlementation de la recherche biomédicale a connu plusieurs modifications au cours des quarante dernières années. La loi Huriet Sérusclat, votée en 1988, a été remplacée par la loi de Santé Publique, votée en 2004, qui définissait deux classes de recherche. Une qui s’intéressait à la recherche d’évaluation des « soins courants » et une qui concernait la recherche « non-interventionnelle » .
La notion de « soins courants » étant considérée comme trop large, la loi Jardé, votée le 5 mars 2012, a permis de préciser l’encadrement de « la recherche impliquant la personne humaine » (RIPH). Les recherches interventionnelles et non-interventionnelles sont classées en trois catégories en fonction du niveau de risque et du degré d’intervention pour les participants. La dernière mise à jour de cette loi est marquée par deux arrêtés mis en application en avril 2018 permettant de préciser encore un peu plus les catégories de recherche définies par cette loi .
La catégorie 1 définit les recherches interventionnelles qui sortent du cadre du soin courant, c’est-à-dire dont l’intervention modifie la prise en charge des patients (que ce soit l’étude d’un médicament expérimental ou d’un acte de soin expérimental). Une assurance souscrite par le promoteur pour les personnes participant à ces recherches est obligatoire.
La catégorie 2 est celle correspondant aux « recherches interventionnelles à risques et contraintes minimes ». C’est pour cette catégorie que les arrêtés de 2018 listent précisément les soins relevant de cette catégorie. Il s’agit de soins non-médicamenteux dont la contrainte est minime pour les patients (une prise de sang dédiée à la recherche par exemple). Une assurance est également obligatoire.
La catégorie 3 correspond aux études non-interventionnelles, anciennement dénommées « observationnelles prospectives », et définit des recherches qui ne modifient pas la prise en charge des participants.
Quelle que soit la catégorie, l’obtention des accords d’un Comité de Protection des Personnes (CPP) et de la Commission National de l’Informatique et des Libertés (CNIL) sont indispensables. Enfin, les études de catégorie 1 doivent obtenir l’accord de l’Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) ; les deux autres catégories ne sont tenues qu’à l’informer de l’étude.
Définitions de principes statistiques
Les définitions suivantes permettront la lecture simplifiée de la suite de ce travail.
1. La validité interne d’une étude est la validité méthodologique de ses résultats, autrement dit, la fiabilité de ses résultats, et donc savoir si les changements observés peuvent être attribués à l’intervention (c’est-à-dire la cause) et non à d’autres causes possibles (« explications alternatives » du résultat). Elle repose sur deux principes : la limitation de biais dans l’étude et la significativité des résultats .
2. La validité externe, elle, correspond au fait de pouvoir appliquer les résultats d’une étude, en pratique.
3. Le biais de sélection est un biais qui affecte une étude au moment de la constitution de l’échantillon de population d’une étude. Il affecte plus précisément le processus par lequel les sujets de l’étude sont inclus.
4. L’analyse en intention de traiter consiste à analyser les résultats des participants dans le groupe dans lequel les patients ont été affectés initialement, et ce, quel que soit le traitement reçu en réalité. Cela permet de préserver les bénéfices la randomisation effectuée.
5. L’analyse per protocole est le contre-pied de l’analyse en intention de traiter. Les patients seront analysés dans le groupe du traitement qu’ils auront effectivement reçu, quel que soit le bras dans lequel ils avaient été randomisés .
Lors de la réalisation d’une étude épidémiologique ou d’un essai clinique, les différences qui seront observées seront soit dues à une véritable différence existante entre les populations étudiées, soit dues au hasard.
6. Le risque alpha représente le risque de conclure à tort à une différence entre deux groupes alors que cette différence est due au hasard ; il est aussi appelé « risque de première espèce ».
7. Le risque beta, lui, représente le risque de ne pas conclure à une différence entre les groupes étudiés alors qu’il existe bel et bien une différence entre les populations dont sont issus les groupes ; il est aussi appelé « risque de deuxième espèce ».
|
Table des matières
INTRODUCTION
PREMIERE PARTIE, GRANDS PRINCIPES DE LA RECHERCHE CLINIQUE
I – HISTORIQUE DE LA RECHERCHE CLINIQUE
II – ENCADREMENT LEGAL DE LA RECHERCHE
III – DEFINITIONS DE PRINCIPES STATISTIQUES
IV – DEFINITION D’UN ESSAI CLINIQUE RANDOMISE (ECR)
Essai de supériorité
Essai de non-infériorité
Essai d’équivalence
V – CRITERES DE QUALITE D’UN ECR
V – RECOMMANDATIONS DE LA HAUTE AUTORITE DE SANTE
DEUXIEME PARTIE : QUELLES SONT LES DIFFICULTES A MENER UN ESSAI CLINIQUE RANDOMISE EN ORTHOPEDIE ? REVUE DE LA LITTERATURE
INTRODUCTION
METHODE
RESULTATS
GENERALITES
CONCEPTION DE L’ESSAI
1. La question à traiter
1.1. Pertinence
1.2. Ethique
1.3. Faisabilité : une étude pilote est-elle nécessaire ?
2. La logistique
2.1. Financement
2.2. Centralisation des données
2.3. Optimiser le recrutement
2.3.1. Au travers de l’équipe soignante
2.3.2. Au travers des patients
RÉALISATION DE L’ESSAI
1. Type d’ECR à réaliser
2. Critère de jugement
3. Nombre de Sujets Nécessaires et taille de l’échantillon
4. Randomisation
5. Gestion de l’insu
6. Gestion des complications
7. Gestion du suivi et des PDV
RÉDACTION ET QUALITÉ DE L’ESSAI
1. Évaluation de la qualité des ECR en orthopédie
1.1. Par la mesure de l’indice de fragilité
1.2. Qualité globale des ECR en orthopédie
2. Rédaction selon les recommandations CONSORT
3. Impact scientifique des ECR en orthopédie
DISCUSSION
GENERALITES
CONCEPTION DE L’ESSAI
1. La question à traiter
1.1. Pertinence
1.2. Ethique
1.3. Faisabilité : une étude pilote est-elle nécessaire ?
2. La logistique
2.1. Financement
2.2. Centralisation des données
2.3. Optimiser le recrutement
2.3.1. Au travers de l’équipe soignante
2.3.2. Au travers des patients
RÉALISATION DE L’ESSAI
1. Type d’ECR à réaliser
2. Critère de jugement
3. Nombre de Sujets Nécessaires et taille de l’échantillon
4. Randomisation
5. Gestion de l’insu
6. Gestion des complications
7. Gestion du suivi et des PDV
RÉDACTION ET QUALITÉ DE L’ESSAI
1. Évaluation de la qualité des ECR en orthopédie
1.1. Par la mesure de l’indice de fragilité
1.2. Qualité globale des ECR en orthopédie
2. Rédaction selon les recommandations CONSORT
3. Impact scientifique des ECR en orthopédie
CONCLUSION
ANNEXE